
2022年11月16日,理论与计算分子科学领域的权威综述期刊WIREs Computational Molecular Science杂志在线发表了中国科学技术大学化学物理系蒋彬课题组题为Atomistic neural network representations for chemical dynamics simulations of molecular, condensed phase, and interfacial systems: Efficiency, representability, and generalization”的受邀综述论文。
机器学习已经广泛应用于化学、物理、生物和材料科学等许多领域,可以通过对量子化学数据的学习,精确预测各种物理和化学性质。其中,原子神经网络(AtNN)方法被广泛应用于跨越不同尺度体系的物理化学尤其是化学动力学研究中。这类方法将体系性质拆分为每个原子的贡献,计算量随总原子数目线性增加,并且可以满足分子、凝聚相和界面体系的不同对称性及周期性,进而实现复杂体系精确且高效的分子动力学模拟。
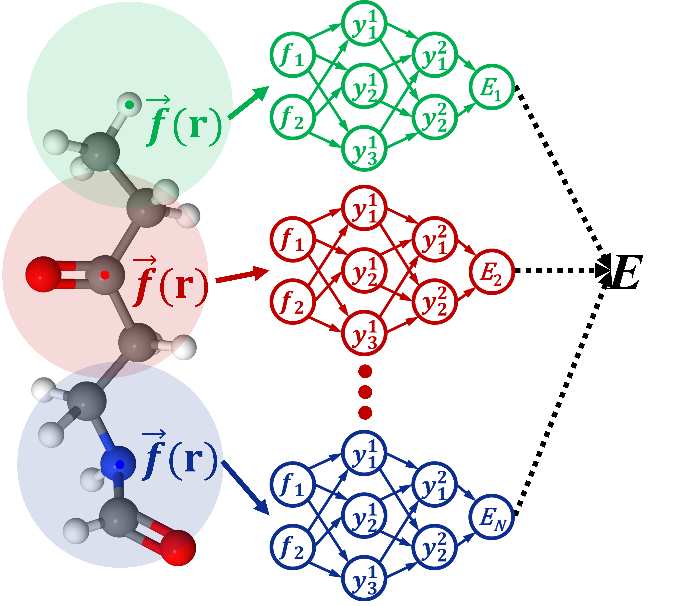
图1:原子神经网络框架示意图。
蒋彬教授课题组从2017年起在AtNN势能面和化学性质预测方面发表了一系列工作。在这篇综述中,蒋彬教授课题组介绍了AtNN模型的基本概念和其背后的物理思想,全面考察了现有各种AtNN方法,以课题组发展的方法为牵引,分类讨论了各种AtNN方法中提升原子环境描述效率和表达力的策略。在效率方面,可以利用各种算法间接计算原子环境中的三体关联,实现对于环境中近邻原子数线性标度,代表方法有蒋彬教授提出的嵌入原子神经网络(EANN),其使用高斯原子轨道线性组合的平方来计算三体关联,显著提升了计算效率。在表达力方面,可以使用多个径向函数的组合来增强两体关联,例如,使用多个高斯原子轨道线性组合作为EANN方法的基函数;更进一步可以使用消息传递网络的形式,递归的引入环境依赖的信息,进而高效地计算高阶多体关联,并引入原子环境外的非局域相互作用,相较于完全基于局域多体描述符的AtNN可以显著提升模型的精度。论文中将不同课题组提出的方法归纳总结到几类策略中,为进一步提升AtNN方法的精度和效率提供了思路。
此外,该论文还总结了将AtNN表达标量势能的模型推广用于表示张量的物理化学性质方面的近期工作。这些工作主要致力于对AtNN置换不变的输出进行张量化处理,以满足张量性质转动协变的对称性。例如,使用坐标和梯度引入方向性质,构建一阶或者二阶张量来表示(跃迁)偶极矩和极化率等响应和跃迁性质;更进一步基于相似的张量化处理的策略,各种AtNN模型可以用来表达电子密度,电子摩擦张量,电子哈密顿量等更为复杂的张量物理性质,并取得了很好的进展。
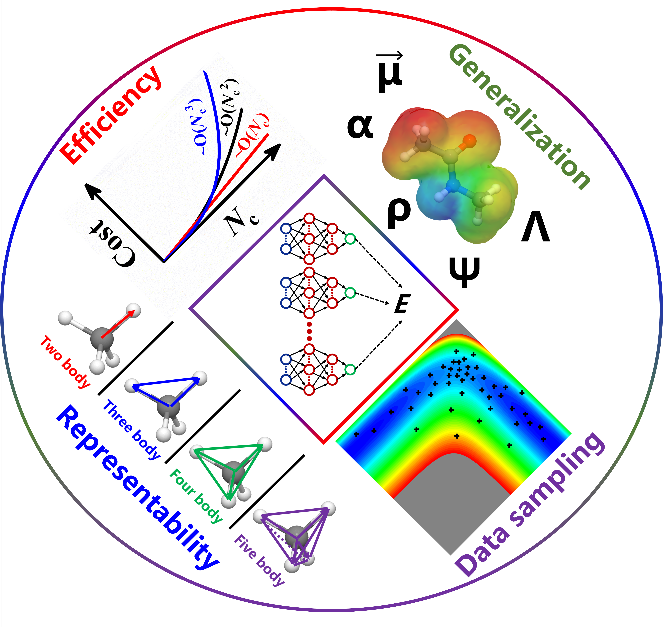
图2:示意图展示原子神经网络表示的主要关注点。
AtNN模型是一个有监督的模型算法,数据对于构建具体体系的模型至关重要。该综述介绍了通过动力学轨线和课题组开发的误差面搜索等主动学习算法,寻找模型不确度较大的区域(往往是数据集缺失的区域),在这个区域上采样新的构型加入数据集,改善模型训练直至达到设定的收敛要求。同时强调了辨别数据相似性的重要性,指出了其难点在于如何高效地考虑原子置换对于构型相似性的影响。论文给出了几个AtNN在气相表面体系中的应用实例,以证明其已经广泛应用于分子和凝聚相体系的化学动力学和光谱模拟。
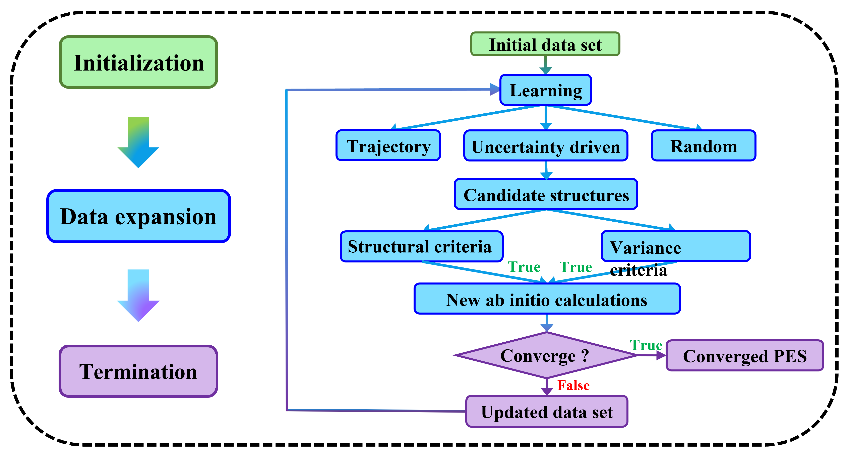
图3:主动学习的流程图。
综上,这篇综述总结了多种策略,旨在构建更高效、更准确、更少人为干预和更通用的机器学习模型,精确表达从电子结构计算中获得的势能和相关物理化学性质。论文最后展望了AtNN方法的发展前景和面临的挑战,指出其有望被进一步推广以表达长程相互作用、外场下的化学性质以及求解电子和原子核薛定谔方程。
中国科学技术大博士生张耀龙和林启东的第一作者和第二作者,蒋彬教授为通讯作者。该工作得到了中科院基础研究青年团队、国家基金委重点项目、安徽省引导性项目、中国科学技术大学创新团队等项目支持。
论文链接:https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1645

