
在硫基电池体系中,引入电催化剂被认为是加速硫还原反应(SRR)的有效途径。然而,硫及其反应中间体(各类硫化物)之间的电催化转化机理仍不够清晰,相关结论尚存争议。原子分散催化剂(ADCs)因金属原子利用率接近100%且具备优异催化活性而备受关注;其活性位点通常集中于中心金属原子及其配位原子,结构清楚,因而更有利于开展精确的构效关系研究。基于这一优势,中国科学技术大学季恒星教授团队长期聚焦金属活性位点的构筑与配位环境优化,提出了高效硫正极催化剂的设计新思路,并在催化位点结构演变、反应机理解析与催化材料理性设计等方面取得一系列进展。
早在2019年,季恒星教授团队就关注到了石墨烯负载的钴单原子在多电子硫反应中的催化作用。通过原位同步辐射X射线吸收谱等先进表征手段,揭示了钴单原子可作为活性位点,有效促进多硫化锂向Li2S的快速转化(J. Am. Chem. Soc.2019, 141, 3977-3985)。随后,团队进一步研究了Co基活性中心在反应过程中的价态演化,发现其与硫化钴物种的动态形成密切相关,可有效改善铝硫电池的电压极化与循环稳定性(Angew. Chem. Int. Ed. 2020, 59, 22963-22967)。在对单原子催化剂的电子结构本质进行深入探究时,研究人员通过实验与理论结合揭示了不同种类金属原子与硫原子之间的d-p轨道匹配对于催化性能的决定性作用(Small2022, 18, 2200395)。基于这一认识,该团队提出了双原子催化位点设计策略,通过Cu与Pt原子间的d轨道耦合调控Pt的d带中心位置,从而增强Pt对硫化物中间体的吸附能力,成功打破线性标度关系并显著提升催化活性(eScience2023, 4, 100222)。此外,针对碳载体在传统认知中仅作为电子导体和催化剂载体的局限性,申请人发现了碳载体的杂化形式影响硫催化活性的载体效应(Angew. Chem. Int. Ed.2023, 62, e202214351)。研究表明碳载体中邻近催化活性中心的sp2杂化碳原子能够提升局域电荷密度,从而增强多硫化物与活性中心之间的电荷转移效率。2024年,该团队进一步提出了活性中心金属原子数量与硫催化活性之间的“火山型”关系,揭示了金属原子数目对催化性能的原子尺度敏感性(J. Am. Chem. Soc.2024, 146, 13055-13065)。在此基础上,研究者优化了活性中心的配位环境,发现金属中心周围氮原子数量的变化对多硫化物中间体的吸脱附行为具有显著影响。当氮配位数由3个减少至1个时,多硫化物吸附行为由不可逆转变为可逆,显著抑制了催化剂中毒并提升循环稳定性(Adv. Funct. Mater.2025, 35, 2501600)。同时,通过掺杂P原子打破Ni单原子配位几何与电子结构的对称性,研究者调控了电子密度分布和中间体吸附能,从而改变硫氧化还原反应动力学,实现了由传统浓度依赖型(一级反应)向表面饱和型(零级反应)的根本转变(Angew. Chem. Int. Ed.2025, 64, e202510212)。这些工作充分表明,催化剂的结构对硫还原活性具有关键影响。因此,如何精准地识别催化剂结构,是深入理解电催化硫还原反应机理并设计高活性硫基电池电催化剂的重要前提。
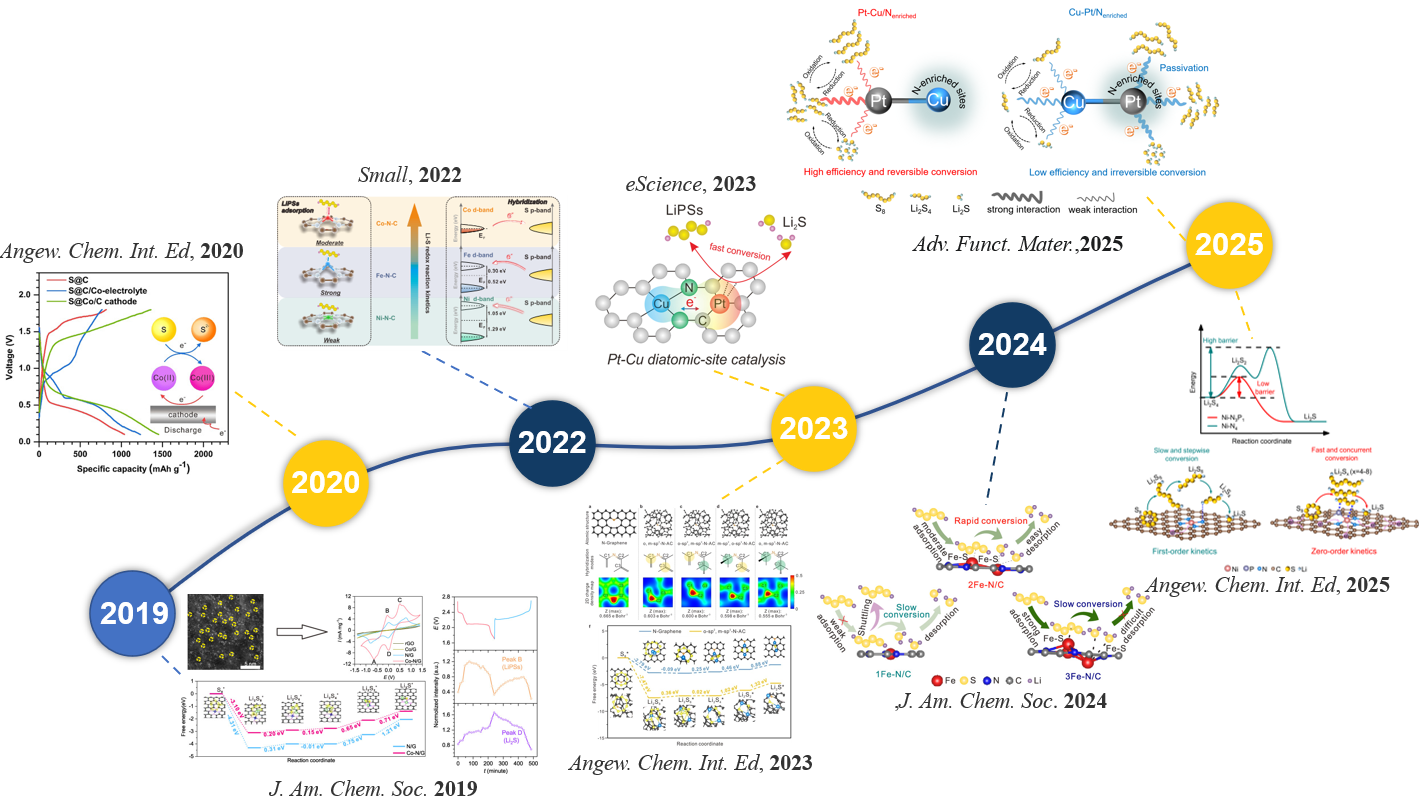
图1 展示了季恒星教授团队近年来在电催化剂研究方面的进展概览。
然而,在实际体系中,载体的非均一性常导致活性中心配位环境的多样化,使得从复杂局部结构中识别具有代表性的结构模型成为理解反应机理的重大挑战。针对这一难题,季恒星教授团队近日提出了一套系统方法,用于识别ADCs的代表性结构模型,涵盖单原子、双原子及三原子催化剂。这一方法有效克服了现实体系中固有的结构异质性及传统EXAFS解析的主观性难题,特别在区分C、N等轻元素配位时展现出显著优势。相关成果以“Identifying Representative Structural Model in Atomically Dispersed Catalysts”为题,于9月25日发表在《美国化学会杂志》(JACS)。
图2 展示了为电催化过程的理论分析提供原子尺度催化中心代表性结构模型的重要性。
X射线吸收谱(XAS)是解析ADCs结构的核心工具,能够提供中心金属原子的平均配位数和价态信息。但由于C、N等轻元素散射能力相近,其配位结构往往难以区分,导致相似的XAS光谱可能对应完全不同的结构模型。其他表征方法如X射线发射谱(XES)、球差校正电镜(STEM)和固体核磁共振(NMR)虽各具优势,但受限于定量能力、分辨率或适用范围。相比之下,结合理论模拟的XAS依然是目前最具可操作性的定量手段,但传统的结构判定方式仍存在一定主观性和不确定性。
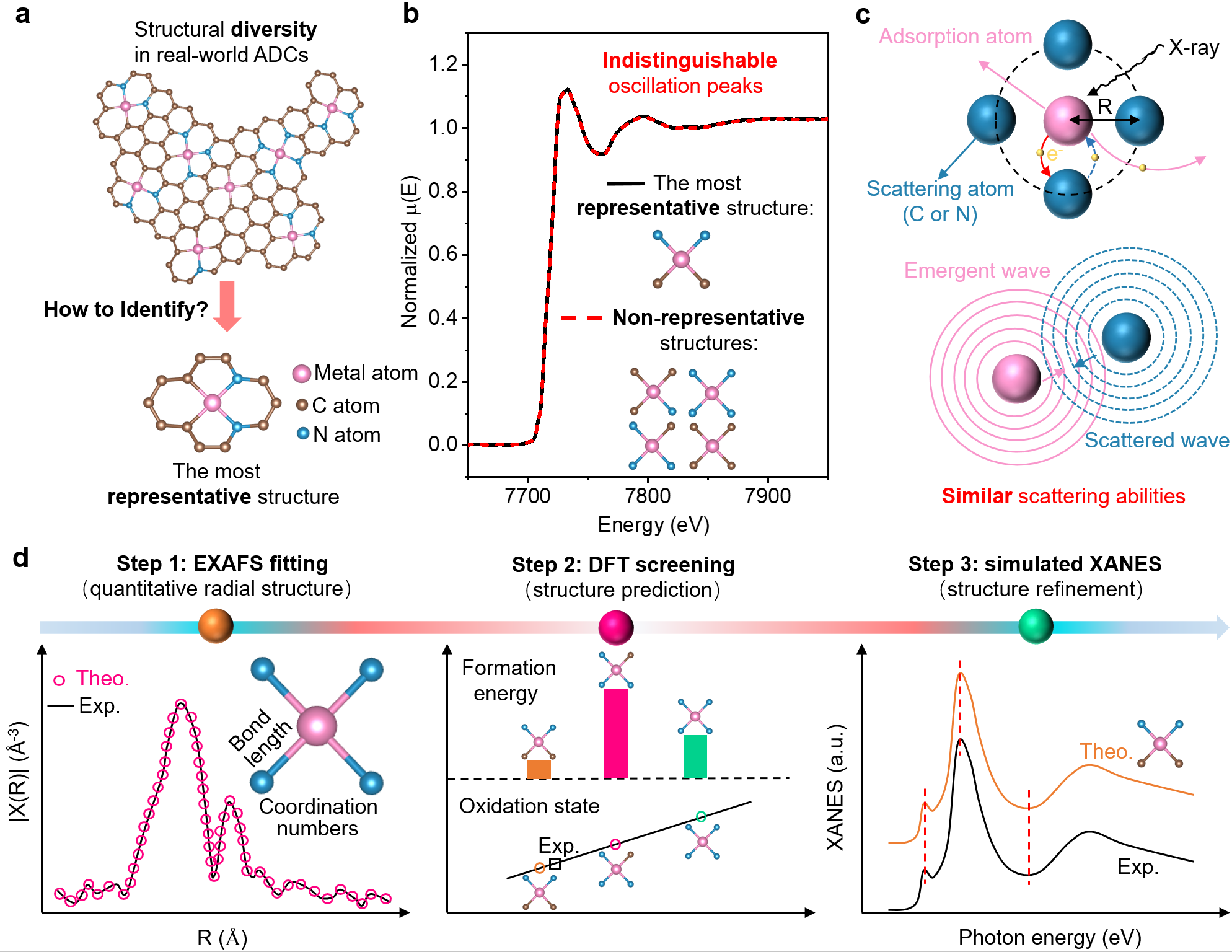
图3 展示了现实ADCs的结构多样性及其代表性结构,并通过XAS谱对比揭示了不同结构对光谱信号的影响及其在区分C、N配位时的局限性。
季恒星教授团队的研究结合了EXAFS与XANES分析,以获取金属中心的配位环境与氧化态信息,并通过DFT计算评估候选结构的热力学稳定性和电子特性。通过将计算与实验XANES谱进行比对,可识别最符合实际的代表性结构,并综合考虑平均配位数、氧化态及配位轻元素的类型,从而显著提升XAS结构解析的可靠性。对于合成过程中可能保留配体的ADCs,研究团队提出了配体敏感流程:首先利用原位红外、DRIFTS或在线TGA-MS定量配体/金属比;随后在EXAFS拟合中保留能够降低R因子且物理合理的配体模式;再通过DFT评估其热力学可行性;最后结合XANES拟合确认吸收边位置、白线强度及近边特征的一致性。整体而言,该研究强调多方法协同在精确识别ADCs活性位点结构中的重要作用,为构效关系的揭示与催化剂理性设计提供了更为可靠的策略。
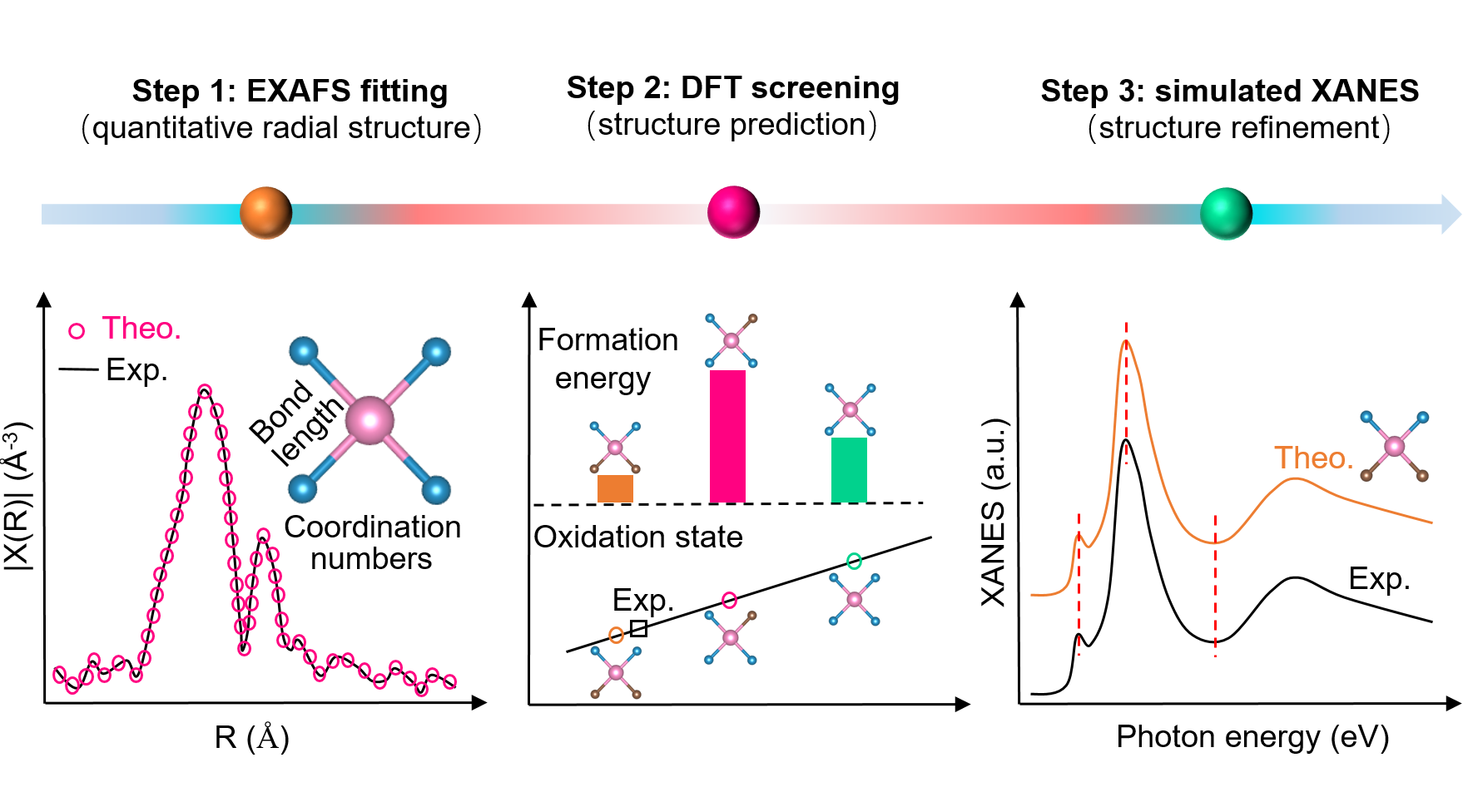
图4 概述了本研究建立的ADCs代表性结构模型识别流程,涵盖候选结构构建、配位环境解析、XAS谱拟合及DFT计算验证,提出了系统化的结构解析策略。
该论文的共同第一作者为中国科学技术大学博士蔡国磊、吕海峰副研究员,通讯作者为季恒星教授和金松特任副研究员。此项工作得到了国家自然科学基金委、中国科学院等项目的支持。
论文链接:https://doi.org/10.1021/jacs.5c09614

