
中国科学技术大学蒋彬教授课题组在发展场诱导的原子神经网络力场研究方面取得重要进展。研究成果以“描述原子体系对于外场响应的通用机器学习力场(Universal machine learning for the response of atomistic systems to external fields)”为题,于2023年10月12日发表在《自然通讯》(Nature Communications)上。
原子模拟是人们在微观层面理解复杂化学、生物和材料体系的光谱、反应动力学以及能量和电荷转移过程的关键工具,其关键要素是精确且高效的高维势能面(即力场)。近年来,基于精确的原子中心的机器学习相互作用势进行高效、准确的分子模拟已经成为一种常用的做法。然而,这些模型通常是用于描述孤立体系,将能量仅视为原子坐标和原子种类的函数,无法表达外场与体系之间的相互作用。外场可以通过与原子、分子或固体材料的相互作用来引发体系的电子极化,自旋极化以及空间取向的变化。这为改变化学结构、控制材料的相变、精确操纵催化反应中的化学反应性和选择性提供了一个重要的工具。因此,我们亟需开发正确描述外场与体系相互作用的机器学习模型,以实现外场下复杂反应的精确且高效的模拟。
蒋彬教授的课题组长期以来一直致力于高精度机器学习力场方法的研究。受到量子化学中原子轨道线性组合为分子轨道的概念启发,研究人员提出了递归嵌入原子电子密度描述符,再将外场视为虚拟的原子(如图1),引入场依赖的原子轨道与基于坐标的原子轨道线性组合来得到对称性适配的场依赖嵌入电荷密度,从而发展出了场诱导的对称性匹配的递归嵌入原子神经网络方法(FIREANN)。该方法能够将偶极矩、极化率等各种响应性质与外场依赖的能量变化精确的关联起来,适用于外场存在下的分子和周期性体系的光谱和动力学模拟。特别值得一提的是,对于周期性体系,这一模型只需训练原子力数据能克服周期性体系内在的极化多值问题。通过甲基乙酰胺和液态水的动力学模拟结果(如图2和图3),验证了这一模型在强外场条件下对各种复杂体系高效建模的能力。
这项研究工作将物理概念与机器学习描述符相关联,为发展更通用的机器学习模型提供了新的思路。

图1:场诱导递归嵌入原子神经网络模型示意图
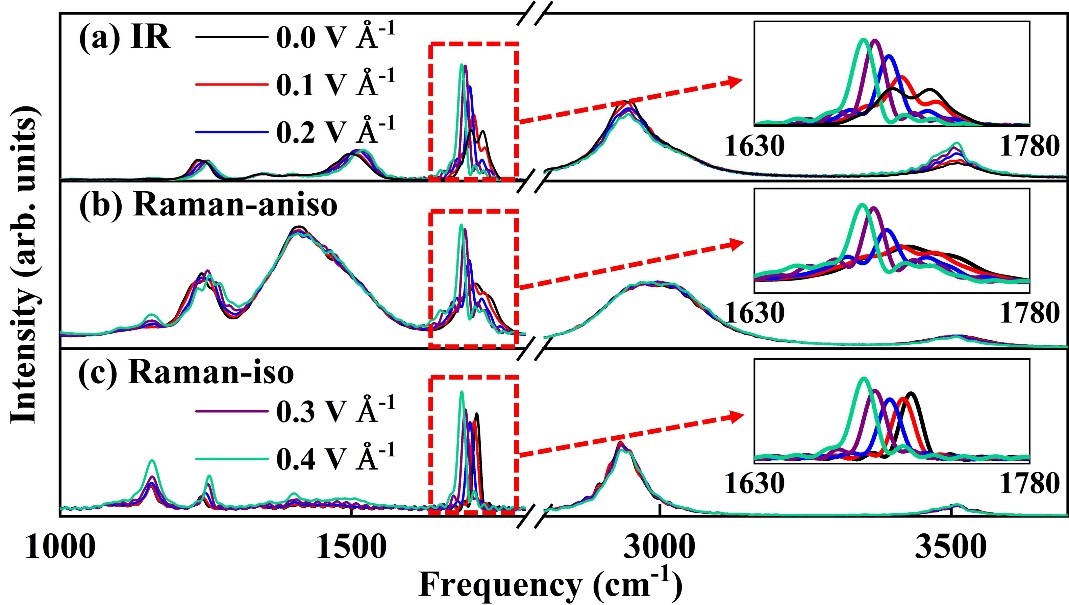
图2:甲基乙酰胺分子的振动光谱
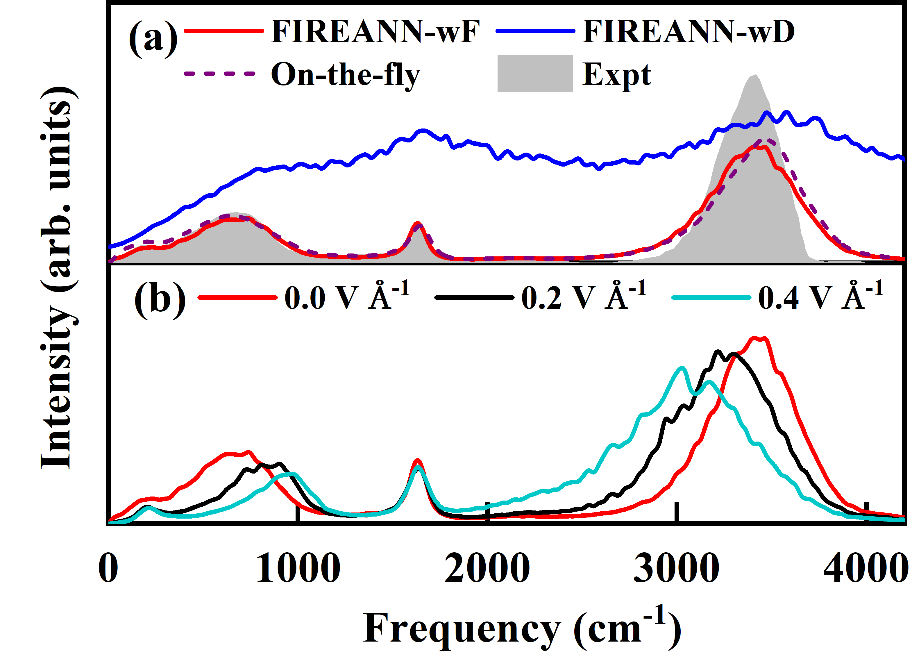
图3:液态水的红外光谱
张耀龙为该论文的第一作者,蒋彬教授为通讯作者。该工作得到了中国科学院战略性先导科技专项、量子科学与技术创新项目、中国科学院稳定支持基础研究领域青年团队、基金委创新群体、重点项目等基金的资助。
论文链接:https://www.nature.com/articles/s41467-023-42148-y

